Installing ChemRender
To help you get started we have a whole page on how to install ChemRender.
To help you get started we have a whole page on how to install ChemRender.
If you get message link the on below it means that you need to enable web linked content.
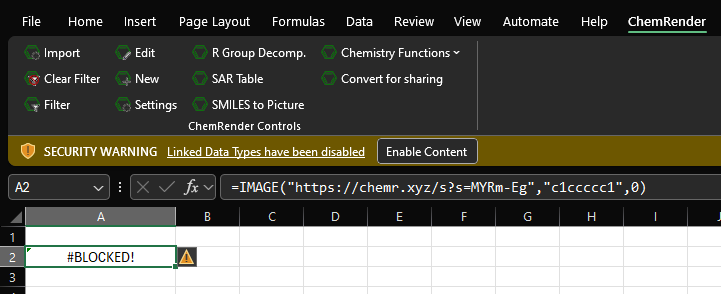
Click the "Enable Content" button will allow the structures to appear.
You may have to re-run the function in the cell, to do this click the cell, then click the Formula bar, before finally pressing enter.
CR.structure(<cell containing a SMILES>)
Renders the smiles as a chemical structure diagram.
CR.formalcharge(<cell containing a SMILES>)
Returns the formal charge.
CR.molarrefractivity(<cell containing a SMILES>)
Returns the molar refractivity.
CR.logp(<cell containing a SMILES>)
Returns the logP value using Crippen's method.
CR.logd(<cell containing a SMILES>)
Returns the predicted CPlogD from a machine learning model (configured to return 80% confidence, option to be configurable in future releases).
The service used can be found at OpenRiskNet.
The supporting paper by Lapins M, Arvidsson S, Lampa S, Berg A, Schaal W, Alvarsson J, Spjuth O can be accessed at doi: 10.1186/s13321-018-0271-1.
CR.molecularweight(<cell containing a SMILES>)
Returns the molecular weight.
CR.exactmolecularweight(<cell containing a SMILES>)
Returns the exact molecular weight.
CR.heavyatommolecularweight(<cell containing a SMILES>)
Returns the heavy atom molecular weight.
CR.fpmorgandensity1(<cell containing a SMILES>)
Returns fp morgan density 1.
CR.fpmorgandensity2(<cell containing a SMILES>)
Returns fp morgan density 2.
CR.fpmorgandensity3(<cell containing a SMILES>)
Returns fp morgan density 3.
CR.maxabspartialcharge(<cell containing a SMILES>)
Returns the maximum absolute partial charge.
CR.maxpartialcharge(<cell containing a SMILES>)
Returns the maximum partial charge.
CR.minabspartialcharge(<cell containing a SMILES>)
Returns the minimum absolute partial charge.
CR.minpartialcharge(<cell containing a SMILES>)
Returns the minimum partial charge.
CR.numradicalelectrons(<cell containing a SMILES>)
Returns the number of radical electrons.
CR.numvalenceelectrons(<cell containing a SMILES>)
Returns the number of valence electrons.
CR.fraction_csp3(<cell containing a SMILES>)
Returns the percentage of sp3-hybridized carbon atoms
CR.heavy_atom_count(<cell containing a SMILES>)
Returns the heavy atom count
CR.num_aliphatic_carbocycles(<cell containing a SMILES>)
Returns the number aliphatic carbocycles
CR.num_aliphatic_heterocycles(<cell containing a SMILES>)
Returns the number of aliphatic heterocycles
CR.num_aliphatic_rings(<cell containing a SMILES>)
Returns the number of aliphatic rings
CR.num_aromatic_carbocycles(<cell containing a SMILES>)
Returns the number aromatic carbocycles
CR.num_aromatic_heterocycles(<cell containing a SMILES>)
Returns the number of aromatic heterocycles
CR.num_aromatic_rings(<cell containing a SMILES>)
Returns the number of aromatic rings
CR.num_h_acceptors(<cell containing a SMILES>)
Returns the number of hydrogen bond acceptors
CR.num_h_donors(<cell containing a SMILES>)
Returns the number of hydrogen bond donors
CR.num_heteroatoms(<cell containing a SMILES>)
Returns the number of heteroatoms
CR.num_rotatable_bonds(<cell containing a SMILES>)
Returns the number of rotatable bonds
CR.num_saturated_carbocycles(<cell containing a SMILES>)
Returns the number of saturated carbocycles
CR.num_saturated_heterocycles(<cell containing a SMILES>)
Returns the number of saturated heterocycles
CR.num_saturated_rings(<cell containing a SMILES>)
Returns the number of saturated rings
CR.ring_count(<cell containing a SMILES>)
Returns the number of rings
CR.num_bonds(<cell containing a SMILES>)
Returns the number of bonds
CR.topographical_polar_surface_area(<cell containing a SMILES>)
Returns the topographical polar surface area
CR.chi0n(<cell containing a SMILES>)
Returns the Chi0n Descriptor
CR.chi0v(<cell containing a SMILES>)
Returns the Chi0v descriptor
CR.chi1n(<cell containing a SMILES>)
Returns the Chi1n Descriptor
CR.chi1v(<cell containing a SMILES>)
Returns the Chi1v descriptor
CR.chi2n(<cell containing a SMILES>)
Returns the Chi2n Descriptor
CR.chi2v(<cell containing a SMILES>)
Returns the Chi2v descriptor
CR.chi3n(<cell containing a SMILES>)
Returns the Chi3n Descriptor
CR.chi3v(<cell containing a SMILES>)
Returns the Chi3v descriptor
CR.chi4n(<cell containing a SMILES>)
Returns the Chi4n Descriptor
CR.chi4v(<cell containing a SMILES>)
Returns the Chi4v descriptor
CR.hallKierAlpha(<cell containing a SMILES>)
Return the Hall Keir Alpha of a molecule
CR.kappa1(<cell containing a SMILES>)
Returns the Hall-Kier Kappa1 Value
CR.kappa2(<cell containing a SMILES>)
Returns the Hall-Kier Kappa2 Value
CR.kappa3(<cell containing a SMILES>)
Returns the Hall-Kier Kappa3 Value
CR.labuteASA(<cell containing a SMILES>)
Returns Labute's Approximate Surface Area (ASA from MOE)
CR.phi(<cell containing a SMILES>)
Returns the PHI Descriptor
CR.lipinskiruleof5(<cell containing a SMILES>)
Returns true or false base on the rules
CR.ghose(<cell containing a SMILES>)
Returns true or false base on the rules
CR.veber(<cell containing a SMILES>)
Returns true or false base on the rules
CR.ruleof3(<cell containing a SMILES>)
Returns true or false base on the rules
CR.reos(<cell containing a SMILES>)
Returns true or false base on the rules
CR.druglike(<cell containing a SMILES>)
Returns true or false base on the rules
CR.smilesfromname(<cell containing a SMILES>)
Returns the smiles for the name provided
CR.iupacnamefromsmiles(<cell containing a SMILES>)
Returns the IUPAC name from the SMILES name provided
CR.inchi(<cell containing SMILES>)
Returns the Inchi for the compound.
CR.inchikey(<cell containing SMILES>)
Returns the Inchi Key for the compound.
CR.molblock(<cell containing SMILES>)
Returns the MOL Block for the compound.
CR.kekule_form(<cell containing SMILES>)
Returns the Kekule Form for the compound.
CR.aromatic_form(<cell containing SMILES>)
Returns the Aromatic Form for the compound.
CR.v3Kmolblock(<cell containing SMILES>)
Returns the V3K MOL Block for the compound.
CR.morgan_fp(<cell containing SMILES>)
Returns the Morgan Fingerprint for the compound.
CR.pattern_fp(<cell containing SMILES>)
Returns the Pattern Fingerprint for the compound.
Please feel free to get in touch using the contact us page.